

Beyond Transcriptomics: RNA Modifications Shape Cancer Progression
RNA modifications such as m6A, A-to-I editing, and pseudouridine influence RNA stability, translation, splicing, and immune recognition. This article explains how these mechanisms shape cancer progression and why they matter for emerging translational applications and therapeutics.


Cancer develops when normal regulatory pathways controlling cell growth, survival, and differentiation become disrupted. Tumor suppressor genes normally restrain proliferation or promote cell death, while oncogenes drive growth and survival. Cancer progression often reflects reduced tumor suppressor activity, increased oncogene signaling, or both.
Historically, these processes have mostly been studied at the level of DNA and transcription, harnessing widely available next-generation sequencing tools. Genetic mutations, copy number changes, and DNA methylation explain many cancer pathologies. However, cancers with similar genomic and transcriptional profiles often diverge markedly in patients, particularly in their response to therapy and their ability to acquire resistance.
This gap highlights that additional regulatory layers influence how genetic information is ultimately expressed. RNA modifications provide a critical layer of regulation that has long been neglected, but is now coming into focus as new technologies enable their detection and quantification. Collectively referred to as epitranscriptomics, RNA modifications are chemical marks added to RNA molecules after transcription. These modifications do not change RNA sequence, but they can alter RNA stability, translation efficiency, splicing decisions, localization, and structure.
From a functional perspective, RNA modifications act downstream of transcription but upstream of protein synthesis. As a result, they can reinforce or reduce oncogenic signaling pathways even when transcriptional levels remain unchanged. They also provide a mechanism for rapid cellular adaptation, as RNA modification states can shift more quickly than transcript or protein turnover and far faster than changes to DNA.
Across cancer biology, the strongest and most consistent evidence reported to date for functional RNA modification dependencies has emerged in hepatocellular carcinoma, myeloid malignancies, prostate cancer, lung cancer, colorectal cancer, glioblastoma, and pediatric cancers such as neuroblastoma. In these settings, m6A methylation, A-to-I RNA editing, and pseudouridine have been mechanistically linked to cancer progression, immune regulation, telomere biology, and therapeutic response.1-9
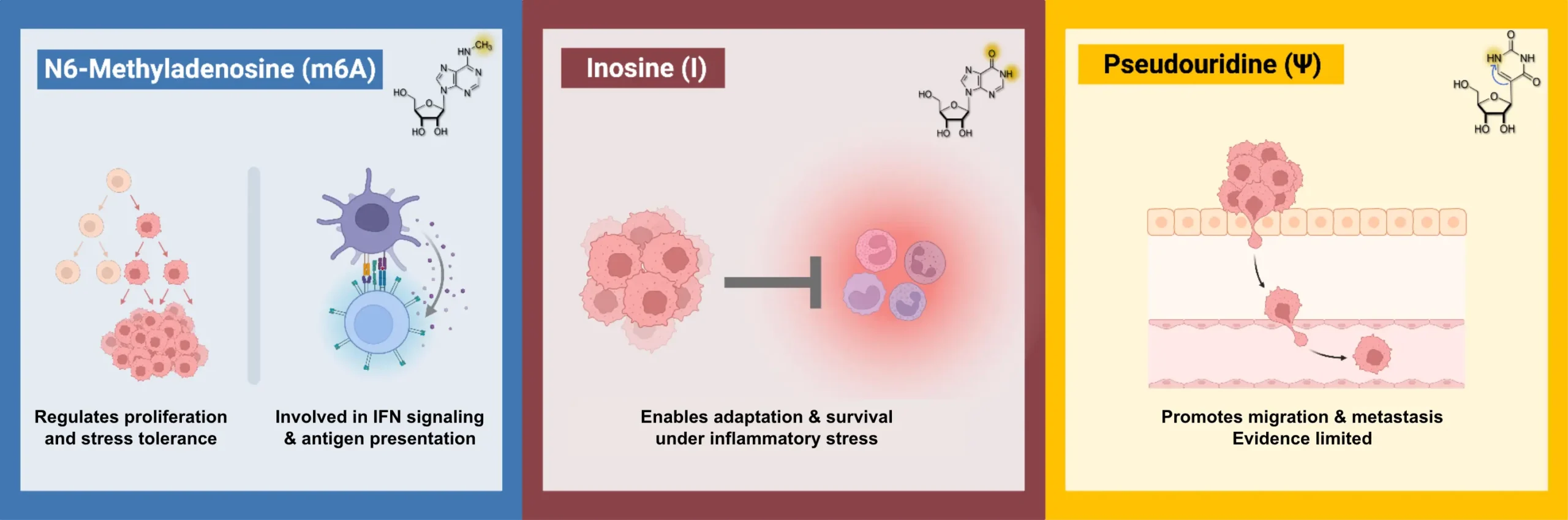
m6A and Cancer Progression
Among RNA modifications, m6A is the most extensively studied in cancer. It is installed by methyltransferases such as METTL3 and METTL14 and erased by demethylases such as FTO and ALKBH5. m6A influences RNA stability, alternative splicing, and translation efficiency by recruiting reader proteins that control RNA fate. Because these processes determine how much functional protein is produced from a given transcript, m6A can reshape signaling pathways without altering transcription.
In hepatocellular carcinoma, METTL3-driven m6A deposition reduces expression of the tumor suppressor SOCS2, leading to sustained oncogenic signaling and increased tumor growth.1 Similarly, in acute myeloid leukemia METTL3 maintains leukemic cell identity by supporting translation of transcripts required for proliferation while blocking differentiation.2 For both of these cases, disrupting m6A writer activity shifts cell state toward less aggressive behavior.
Human prostate cancer provides a clear example of the clinical relevance of this biology. A study profiling m6A patterns across localized prostate tumors identified distinct m6A-defined disease states associated with grade, hypoxia, tumor size, and recurrence risk.4 These states were not fully explained by gene expression alone, indicating that RNA modification patterns capture disease features invisible to standard transcriptomic analysis.
In neuroblastoma, METTL3-dependent m6A modification of the telomeric long noncoding RNA TERRA is required for telomere targeting and maintenance in tumors that use Alternative Lengthening of Telomeres (ALT), demonstrating how RNA modification directly supports telomere biology in pediatric cancer.9
m6A-dependent regulation has also been observed in lung adenocarcinoma, colorectal cancer, and glioblastoma, where it supports stress adaptation, survival signaling, and stem-like programs linked to progression.3,5,6 Collectively, these findings position RNA methylation machinery as a conserved regulator of cancer cell state across diverse tumor types.
m6A, RNA Processing, and Immune Regulation
Beyond translation control, m6A can influence RNA splicing and RNA-protein interactions, contributing to pathway rewiring under stress or therapy. This layer of regulation becomes particularly relevant in cancer, where alternative splicing and altered RNA processing frequently accompany progression and resistance.
m6A also influences cancer-immune interactions. Depletion of METTL3 and METTL14 enhances interferon signaling and antigen presentation, converting tumors previously insensitive to PD-1 blockade into responders.10 Broader analyses show that m6A affects immune checkpoint expression, T cell activation, dendritic cell maturation, and natural killer cell function.11
With increasing mechanistic insights into the role of m6A in immune system function, a promising therapeutic avenue has emerged through catalytic inhibitors of METTL3 that activate anti-cancer immune responses.12 Together, these findings link RNA methylation to both intrinsic cancer signaling and extrinsic immune recognition, providing opportunities for extended clinical monitoring applications and the development of novel anti-cancer drugs.
A-to-I RNA Editing and Innate Immune Sensing
A-to-I RNA editing, primarily mediated by the enzyme ADAR1, serves as a critical checkpoint to prevent “viral mimicry,” a state where the cell’s own RNA triggers a lethal immune response. By converting adenosine to inosine in double-stranded RNA, ADAR1 destabilizes these duplexes, masking “self” RNA from innate sensors such as MDA5 and PKR that would otherwise recognize it as foreign.
In solid tumors, ADAR1 often acts as a mechanism of adaptive resistance. Disabling this axis has been shown to restore sensitivity to immune checkpoint blockade by reactivating dormant sensing pathways.7 This dependency is particularly acute in cancers with high interferon-stimulated gene signatures, where cells become addicted to ADAR1 to survive a state of chronic inflammatory stress.13,14
This interplay between ADAR1 and the immune microenvironment is a key regulator of disease progression, as seen in colorectal cancer where editing levels correlate with immune cell infiltration.15 Similarly, in hematologic malignancies, ADAR1 is essential for leukemia stem cell maintenance. Under inflammation, a malignant switch to the ADAR1p150 isoform suppresses chronic interferon signaling that would otherwise trigger apoptosis.16,17
These findings establish A-to-I editing as a conserved mechanism regulating immune signaling and cancer progression across diverse disease contexts.14 This vulnerability offers significant therapeutic potential. For example, small molecules that reverse malignant isoform switching are being investigated as a strategy to trigger apoptosis in cancer stem cells.18
Pseudouridine and RNA Structure
Pseudouridine stabilizes RNA structure and influences RNA-protein interactions. While long studied in noncoding RNA, recent work highlights its role in messenger RNA and immune regulation.
Loss of the pseudouridine synthase PUS1 activates antiviral signaling and enhances response to PD-1 therapy.8 This suggests that pseudouridine normally limits innate immune recognition by stabilizing RNA structures, and that disrupting this modification exposes RNAs that trigger immune activation. Broader analyses also link pseudouridine to stress responses and RNA stability, supporting its relevance to cancer progression.19
Emerging Translational Applications
RNA modifications respond dynamically to environmental and therapeutic pressures. Because they operate downstream of transcription, they can reveal pathway activity and adaptation that are not captured by gene expression alone.
A large pharmacogenomic analysis across multiple cancer types demonstrated that m6A profiles predict drug sensitivity even when transcript levels fail to distinguish responders from non-responders.20 This work provides direct evidence that RNA modification states carry orthogonal predictive value relative to genomics and transcriptomics.
Together with growing evidence linking RNA modifications to splicing regulation, immune evasion, and telomere maintenance, these findings suggest that RNA modifications may serve as informative biomarkers of cancer state and therapeutic response, and as potential points of intervention for reshaping cancer cell behavior.
Conclusion
Across different cancers including solid tumors, pediatric cancers such as neuroblastoma, and blood cancers including myeloid malignancies, RNA modifications such as m6A, A-to-I editing, and pseudouridine contribute to cancer progression, phenotype, metabolic state, immune regulation, and therapeutic response. Because these pathways cannot be fully captured dynamically by transcriptomics and genomic profiling alone, RNA modification profiling is essential for understanding cancer biology at a fundamental level.
RNA modifications respond dynamically to environmental and therapeutic pressures on timescales that complement and extend those revealed by genomics and transcriptomics. However, broader adoption in translational research has been limited by practical challenges, including the difficulty of measuring modification abundance and precise transcript location at scale, variability across platforms and protocols, limited availability of reference standards, and incomplete understanding of how specific modification patterns translate to functional or clinical outcomes.
Recent advances in profiling technologies are now addressing these constraints, enabling more sensitive and spatially resolved measurement of RNA modifications from limited clinical material, including biopsies and liquid biopsies.* As RNA modification characterization tools continue to mature, integrating these data layers with genomic and transcriptomic information in the multi-omics toolbox holds strong promise for improving clinical strategies and patient outcomes.
Explore additional posts in our Mods in Motion series
Blog Posts
- The Power of Inosine: How RNA Editing Shapes the Transcriptome
- A Beginner’s Guide to m6A and Other RNA Modifications
Podcasts
About the Author
James Tsay
James M. Tsay, PhD, is a biotech scientist and industry leader whose work spans across genomics, molecular diagnostics, and multi-omic technologies. He earned his PhD in physical chemistry at UCLA and completed postdoctoral research in biophysics at UC San Diego. He is currently a Scientific Advisor at AlidaBio, and previously held senior scientific and technical leadership roles at Illumina and Pleno Biosciences, contributing to the development of next-generation sequencing and advanced molecular profiling platforms.
References
- Chen, M., Wei, L., Law, C.-T., Tsang, F.-H. C., et al. (2018). RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through m6A-dependent repression of SOCS2. Hepatology, 67(6), 2254–2270. https://doi.org/10.1002/hep.29614
- Barbieri, I., Tzelepis, K., Pandolfini, L., Shi, J., et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature, 552(7683), 126–131. https://doi.org/10.1038/nature24678
- Xu, Y., He, X., Wang, J., Zhang, Q., et al. (2022). METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 via m6A modification. Cancer Cell International, 22(1), 11. https://doi.org/10.1186/s12935-021-02433-6
- Li, X., Xu, X., Wang, J., Zhang, Y., et al. (2025). Distinct N6-methyladenosine modification patterns define subtypes of prostate cancer with clinical relevance. Nature Genetics. https://doi.org/10.1038/s41588-025-02128-y
- Li, T., Hu, P.-S., Zuo, Z., Lin, J.-F., et al. (2019). METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Molecular Cancer, 18, 112. https://doi.org/10.1186/s12943-019-1038-7
- Cui, Q., Shi, H., Ye, P., Li, L., et al. (2017). m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Reports, 18(11), 2622–2634. https://doi.org/10.1016/j.celrep.2017.02.059
- Ishizuka, J. J., Manguso, R. T., Cheruiyot, C. K., Bi, K., et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature, 565(7737), 43–48. https://doi.org/10.1038/s41586-018-0768-9
- Wang, F., Tong, Y., Guo, W., Qian, Y., et al. (2025). Pseudouridine synthase 1-targeted therapy activates antiviral immunity to boost cancer immunotherapy. Cell Reports, 44(9), 116233. https://doi.org/10.1016/j.celrep.2025.116233
- Vaid, R., Thombare, K., Mondal, T., Singh, A., et al. (2024). METTL3 drives telomere targeting of TERRA lncRNA through m6A-dependent R-loop formation in alternative lengthening of telomeres-positive neuroblastoma. Nucleic Acids Research, 52(5), 2648–2671. https://doi.org/10.1093/nar/gkad1242
- Wang, L., Hui, H., Agrawal, K., Kryczek, I., et al. (2020). METTL3 and METTL14 regulate response to anti-PD-1 immunotherapy. The EMBO Journal, 39(19), e104514. https://pmc.ncbi.nlm.nih.gov/articles/PMC7560214/
- Qin, Y., Zhang, Y., Zhang, L., Li, Z., et al. (2024). RNA modifications and their roles in cancer immunity and immunotherapy. Frontiers in Immunology, 15, 1412345. https://doi.org/10.3389/fimmu.2024.1463847
- Yankova, E., Blackaby, W., Albertella, M., et al. (2021). Small-molecule inhibition of METTL3 as a strategy against acute myeloid leukemia. Nature, 593, 597–601. https://doi.org/10.1038/s41586-021-03536-w
- Gannon, H. S., Zou, T., Kiessling, M. K., Gao, G. F., et al. (2018). Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nature Communications, 9(1), 5450. https://doi.org/10.1038/s41467-018-07824-4
- Liu, H., Golji, J., Brodeur, L. K., Chung, F. S., et al. (2019). Tumor-derived interferon triggers chronic pathway agonism and sensitivity to ADAR loss. Nature Medicine, 25, 95–102. https://doi.org/10.1038/s41591-018-0302-5
- Zheng, G.-L., Zhang, G.-Y., Zhao, Y., Zheng, Z.-C., et al. (2023). The interplay between RNA editing regulator ADAR1 and the immune environment in colorectal cancer. Journal of Oncology, 2023, Article ID 9315027. https://doi.org/10.1155/2023/9315027
- Zipeto, M. A., et al. (2016). ADAR1 activation drives leukemia stem cell self-renewal by impairing let-7 biogenesis. Cell Stem Cell, 19(2), 177–191. https://doi.org/10.1016/j.stem.2016.05.004
- Jiang, Q., et al. (2013). ADAR1 promotes malignant progenitor proliferation in chronic myeloid leukemia. Proceedings of the National Academy of Sciences, 110(3), 1041–1046. https://doi.org/10.1073/pnas.1213021110
- Crews, L. A., Ma, W., Ladel, L., Pham, J., et al. (2023). Reversal of malignant ADAR1 splice isoform switching with Rebecsinib. Cell Stem Cell, 30(3), 250–263.e6. https://doi.org/10.1016/j.stem.2023.01.008
- Jia, S., Yu, X., Deng, N., Gao, Z., et al. (2025). Deciphering the pseudouridine nucleobase modification in human diseases: From molecular mechanisms to clinical perspectives. Clinical and Translational Medicine, 15, e70190. https://doi.org/10.1002/ctm2.70190
- Liu, K., Ouyang, Q.-Y., Zhan, Y., Yin, H., et al. (2022). Pharmacoepitranscriptomic landscape revealing m6A modification could be a drug-effect biomarker for cancer treatment. Molecular Therapy – Nucleic Acids, 28, 464–476. https://doi.org/10.1016/j.omtn.2022.04.001
Additional Technical Resources
- *GEN Eng News (2023). Beyond the map: capturing abundance and location of RNA modifications from ultra-low inputs.Genetic Engineering & Biotechnology News.https://www.genengnews.com/topics/bioprocessing/beyond-the-map-capturing-abundance-and-location-of-rna-modifications-from-ultra-low-inputs/